I have a problem with a bacterial transformation of a yeast gene that I can not solve.
I isolated yeast DNA and did a PCR to get my product. I am using pCGCUm vector with a GFP construct. I digest both my plasmid and my product with SmaI and ClaI for 4h each. After that I purify the product from a gel and perform a ligation with high efficiency T4 ligase for 30 minutes at 22°C. Then I transform my electrocompetent cells with electroporation (2000V, 5ms), recover it in 0.5ml LB for 2h and plate it on LB-A plates.
First time I only did:
- insert1
- insert2
- Empty vector
and it did not work (empty plates for insert and confluent growth (after 18h) of some weird colonies on EV).
I did the same ligation again and this time I included controls
This is what I did:
- insert1
- insert2
- Empty vector
- negative control (EK cells + electroporation)
- positive control (EK cells + undigested vector)
Now I got:
- insert1 (no colonies)
- insert2 (big colonies a lot of them)
- Empty vector (big colonies a lot of them)
- negative control (EK cells + electroporation) (no colonies)
- positive control (EK cells + undigested vector) (big colonies a lot of them)
The negative control suggests that my competent cells are ok, at least they are not resistant to AMP.
What confuses me is that even with positive control I get some weird big colonies - if I got nice transformants here I would assume that either digestion of ligation was not successful, but now I doubt that).
Another thing that is weird is that now I got that colonies also on my insert2 plate.
The scan image of big colonies looks like that:
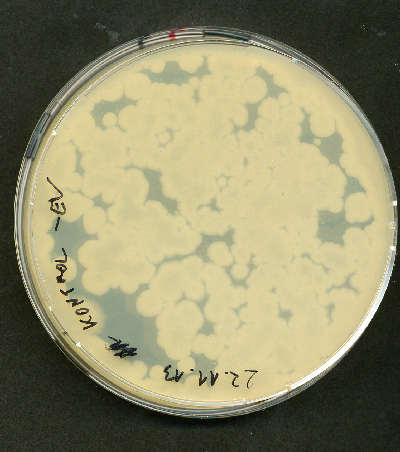
I have a question if anyone has had that kind of colonies? I was thinking of maybe having contaminated cuvettes for electroporation but I had them in 100% EtOH for 3 days and then dried in fume hood for 3 hours before electroporation.