While the heart is affected during septic shock, it is the result of the chain of events that occur as a result of a systemic release of the pro-inflammatory cytokine Tumor Necrosis Factor alpha
(TNF-$\alpha$) that leads to the "collapse of the circulatory and respiratory system." (Janeway's Immunobiology, 8th Edition) See also Tumor necrosis factor signaling.
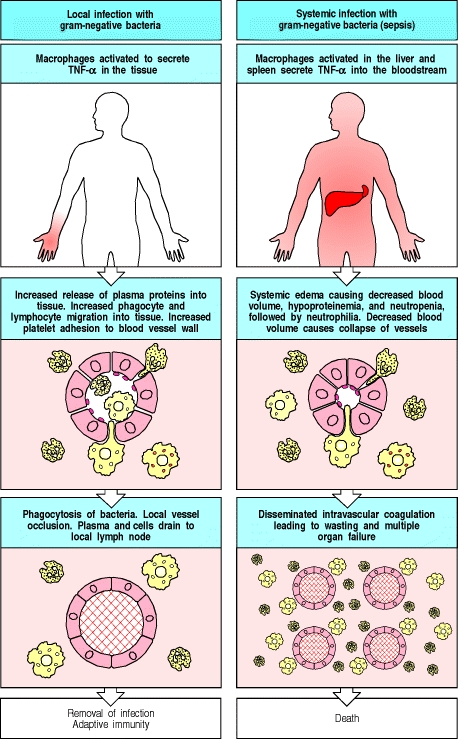
When leukocytes, mainly macrophages encounter a pathogen or the antigenic product of a pathogen, pathogen-associated molecular patterns (PAMPs) such as endotoxins or exotoxins, their pattern recognition receptors (PRRs), such as the toll-like receptors, are bound and signaling occurs which stimulates the production and release of pro-inflammatory cytokines. In a localized infection, this is a beneficial response, however when the response is systemic, it will lead to a catastrophic cascade that leads to septic shock and often death.
Under "normal" conditions, TNF-$\alpha$ is an inducer of local inflammatory response. When an endotoxin such as lipopolysaccharide (LPS) is detected by TLR-4 on macrophages in the local microenvironment of an infection, the macrophages are stimulated to secrete TNF-$\alpha$. TNF-$\alpha$ produces the following physiological effects:
- Activates the vascular endothelium
- The endothelial cells will begin expressing a different assortment of receptors that recruit leukocytes out of the blood and into the tissue
- This also allows for increased platelet adhesion to the activated endothelial vessels
- Increases vascular permeability
- Increased entry of Immunoglobulin G into the infected tissue
- Increases entry of Complement proteins from the plasma to the tissue
- Increased access to leukocytes and activated lymphocytes for extravasation into the infection microenvironment
- Increased fluid drainage to lymph nodes
- Janeway's Immunobiology, 8th Edition; p 101
One of the mechanisms that is beneficial to the local inflammatory response, namely platelet adhesion will become problematic in the systemic response and will lead to multi-organ failure. In the local environment, however, it helps to prevent the spread of infection and shunts fluid flow away from the bloodstream and to the lymphatic system where it will be encountered by the components of the adaptive immune system. Locally:
Another important action of TNF-$\alpha$ is to stimulate endothelial cells to express proteins that trigger blood clotting in the local small vessels, occluding them and cutting off blood flow. This can be important in preventing the pathogen from entering the bloodstream and spreading through the blood to organs all over the body. Instead, the fluid that has leaked into the tissue in the early phases of an infection carries the pathogen, usually enclosed in dendritic cells, via the lymph to the regional lymph nodes, where an adaptive immune response can be initiated.
- Janeway's Immunobiology, 8th Edition; pp 107-108
You can begin to see where the inflammatory response triggered by TNF-$\alpha$ may become a catastrophic problem if it is secreted systemically.
Once an infection spreads to the bloodstream, however, the same mechanisms by which TNF-$\alpha$ so effectively contains local infection instead become catastrophic. Although produced as a membrane associated cytokine, TNF-$\alpha$ can be cleaved by a specific protease TACE (TNF-$\alpha$-converting enzyme, formerly called ADAM17) and released from the membrane as a soluble cytokine. The presence of infection in the bloodstream, or sepsis, is accompanied by a massive release of TNF-$\alpha$ by macrophages in the liver, spleen, and other sites throughout the body. The systemic release of TNF-$\alpha$ into the bloodstream causes vasodilation, which leads to a loss of blood pressure and increased vascular permeability, leading to a loss of plasma volume and eventually shock, known in this case as septic shock because the underlying cause is a bacterial infection. The TNF-$\alpha$ release in septic shock also triggers blood clotting in small vessels throughout the body--known as disseminated intravascular coagulation--which leads to the massive consumption of clotting proteins, so that the patient’s blood cannot clot appropriately. Disseminating intravascular coagulation frequently leads to the failure of vital organs such as the kidneys, liver, heart, and lungs, which are quickly compromised by the failure of normal perfusion; consequently, septic shock has a high mortality rate.
- Janeway's Immunobiology, 8th Edition; pp 108-109
So, as you can see, it is the effect of the depletion of clotting factor, the loss of plasma volume and blood pressure, and the alteration of the blood's ability to clot which likely is leading to the arrhythmic effects that you have observed in the heart.
The figure below summarizes both the localized and systemic effects of TNF-$\alpha$ release.
 $\alpha$ can have damaging effects when released systemically" />
$\alpha$ can have damaging effects when released systemically" />
- Janeway's Immunobiology, 5th Edition; Figure 2.37
Effects of TNF-$\alpha$ on the heart
There is research from the 1990s and early 2000s that deals with the effects of TNF-$\alpha$ both on the heart and expression of the cytokine in failing heart tissue. Below is a brief selection of those articles. The final reference discusses the link between Toll-like receptor 4 activation and the promotion of cardiac arrhythmias:
From Cardiac-specific overexpression of tumor necrosis factor-alpha causes lethal myocarditis in transgenic mice. Kubota, et.al. J Card Fail. 1997 Jun;3(2):117-24.
Tumor necrosis factor (TNF)-alpha, a proinflammatory cytokine with negative inotropic effects, can be detected in myocardium with end-stage heart failure, after endotoxin administration, and during transplant rejection. Various studies suggest that TNF-alpha participates in the pathogenesis of cardiac dysfunction.
Overexpression of TNF-alpha in the heart leads to severe myocarditis and cardiomegaly. These results support the hypothesis that myocardial expression of TNF-alpha can contribute to the pathogenesis of cardiac dysfunction.
From Dilated Cardiomyopathy in Transgenic Mice With Cardiac-Specific Overexpression of Tumor Necrosis Factor-$\alpha$. Kubota, et.al.
Circulation Research.1997; 81: 627-635; doi: 10.1161/01.RES.81.4.627
That end-stage congestive heart failure is associated with a marked increase in circulating TNF-$\alpha$ levels has been reported by numerous investigators. Furthermore, there appears to be a direct relationship between disease severity and circulating levels of TNF-$\alpha$.
In the present study, we demonstrate that chronic overexpression of TNF-$\alpha$ results in the development of a dilated cardiomyopathy that shows (1) ventricular hypertrophy, (2) ventricular dilatation, (3) interstitial infiltrates, (4) interstitial fibrosis, (5) rare myocyte apoptosis, (6) a diminished ejection fraction, (7) attenuation of $\beta$1-adrenergic responsiveness, and (8) expression of atrial natriuretic factor in the ventricle. Additionally, mice overexpressing the TNF-α transgene had a marked increase in mortality. More than half of the mice that died spontaneously presented with exceptional dilatation of the heart, increased lung weight, and pleural effusion, suggesting that they died of congestive heart failure.
From The role of tumor necrosis factor in the pathophysiology of heart failure Feldman, et.al. Journal of the American College of Cardiology
Volume 35, Issue 3, 1 March 2000, Pages 537–544
In summary, both basic and clinical studies strongly support the hypothesis that myocardial expression of TNF alpha is an important step in the pathophysiologic pathway leading to progressive cardiac dilatation and failure.
From Toll-like receptor 4 activation promotes cardiac arrhythmias by decreasing the transient outward potassium current (Ito) through an IRF3-dependent and MyD88-independent pathway. Monnerat-Cahli, et. al. J Mol Cell Cardiol. 2014 Nov;76:116-25. doi: 10.1016/j.yjmcc.2014.08.012. Epub 2014 Aug 25.
Abstract
Cardiac arrhythmias are one of the main causes of death worldwide. Several studies have shown that inflammation plays a key role in different cardiac diseases and Toll-like receptors (TLRs) seem to be involved in cardiac complications. In the present study, we investigated whether the activation of TLR4 induces cardiac electrical remodeling and arrhythmias, and the signaling pathway involved in these effects. Membrane potential was recorded in Wistar rat ventricle. Ca(2+) transients, as well as the L-type Ca(2+) current (ICaL) and the transient outward K(+) current (Ito), were recorded in isolated myocytes after 24 h exposure to the TLR4 agonist, lipopolysaccharide (LPS, 1 μg/ml). TLR4 stimulation in vitro promoted a cardiac electrical remodeling that leads to action potential prolongation associated with arrhythmic events, such as delayed afterdepolarization and triggered activity. After 24 h LPS incubation, Ito amplitude, as well as Kv4.3 and KChIP2 mRNA levels were reduced. The Ito decrease by LPS was prevented by inhibition of interferon regulatory factor 3 (IRF3), but not by inhibition of interleukin-1 receptor-associated kinase 4 (IRAK4) or nuclear factor kappa B (NF-κB). Extrasystolic activity was present in 25% of the cells, but apart from that, Ca(2+) transients and ICaL were not affected by LPS; however, Na(+)/Ca(2+) exchanger (NCX) activity was apparently increased. We conclude that TLR4 activation decreased Ito, which increased AP duration via a MyD88-independent, IRF3-dependent pathway. The longer action potential, associated with enhanced Ca(2+) efflux via NCX, could explain the presence of arrhythmias in the LPS group.