I was wondering if both all b and y ions are required to determine to order of the amio acids in a peptide.
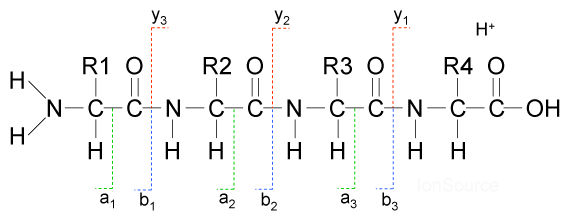
In my opinion knowing only ALL B ions or ALL y-ions is enough to determine the sequence of the amino acids in a peptide. I was also wondering how these b and y ions are differentiated in a spectrum?
$\begingroup$
$\endgroup$
1
-
$\begingroup$ I'm struggling to understand the diagram. You're asking about b and y ions, but b and y are pointing to C-N bonds. Could you expand. I'm not exactly sure what the bioinformatics tag is doing here. $\endgroup$– JamesCommented Oct 10, 2016 at 3:11
Add a comment
|
1 Answer
$\begingroup$
$\endgroup$
In an MS/MS spectrum, difference between the successive peaks of a b or y ion series allows one to determine the amino acids making the series/peptide. Either one of them should be enough to give you the sequence. However, an MS/MS spectrum obtained from the mass spec instrument doesnt tell fragment types. Therefore it takes some expertise and tricks to spot y or b ion series. For example: if you are analysing a sample digested by trypsin, your C-term residue will either be K or R. So you can look for a Y1 ion if there is 147 indicating K or 175 indicating R. Also, Y ions tend to be highest intensity peaks in a spectrum. For more helpful tips on this there is an excellent tutorial : http://www.ionsource.com/tutorial/DeNovo/DeNovoTOC.htm.
Also, in practice you seldom get all of the b or y ion peaks and there are often missing peaks. For this reason it is useful to have both b and y ions series so that gaps in one can be filled by the other. This is not only useful when you are dealing with denovo sequencing but also in a more typical shotgun proteomics experiment. In a typical shotgun proteomics experiment one searches their mass spec data with peptide search engines. These are software which use a protein sequence database which is in silico digested, based on the specificity of the protease used in the experiment, and from the resulting peptide sequences search engines make a library of predicted MS/MS spectra (this is done by fragmenting along the peptide backbone and listing all possible b and y ions). Then these predicted spectra are matched with the observed spectra to predict the peptide sequences that could result in the observed spectra. Again, not all of the peaks present in a predicted spectra will be found in an observed spectra. So it is best to have ions from both b and y ion series. More ions matched between the observed and predicted spectra, better is the score and confidence in identification.