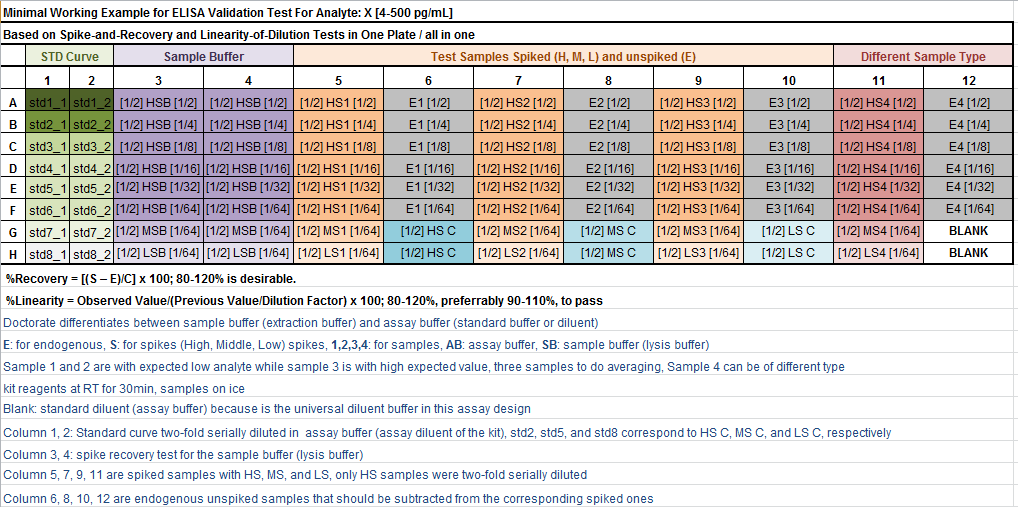
I have searched in the web for a detailed explanation of doing such validation experiment, but unfortunately couldn't find a satisfactory one. I came across the following sources:
Thermo Scientific Tech tip #58: IMAHO, well written description, but still there are some issues
Pros
- Clear on describing terms and straight forward calculations
- Troubleshooting of the results is outlined
Cons
- Not designed to be performed in one 96-well plate ELISA
- No mention of any acceptable criteria for the both validation tests
- No mention of
blank
Quansys Biosciences: a practical design to implement
Pros
- Both spike-and-recovery and linearity-of-dilution experiments can be performed in one 96-well plate ELISA
- Justified introduction of the
endogenous sample, which is diluted the same amount as the spiked sample - Addition of
blank - Mention of acceptance criteria for both types of experiments
Cons
- No mention of troubleshooting
- a little bit messy plate design
R&D spike and recovery protocol: a brief one but useful
Pros
- Clear on design
- Mention of acceptable criteria at least for the spike recovery 80-120%
- Mention of
blank - Hint about possibility of using diluted neat sample until it gives reading
Cons
- One sample was used for assessment and not averaging on 3 samples at least
- No mention for any criteria for linearity of dilution
Issues/Questions
- Q1: How can we combine the two experiments into one plate to spare samples, and materials?
- Q2: Can we dilute sample with assay diluent instead of sample diluent?
- Q3: In case low sample material is there, can we skip the neat sample and add 1:2 diluted one instead to spare material?
- Q4: In preparing spikes in Thermo Scientific tech#58, why was it 10µl spike stock solution added to 50µl sample? what governs this addition, is there any ratio to stick to here or arbitrary ratio? Can we add 90µl sample and 10µl stock solution?
Please feel free to expand sources, or questions, and of course answers are more than welcome to achieve the following aim:
The best design for both validation tests (i.e., spike-and-recovery and linearity-of-dilution) in one 96-well plate ELISA that will use the minimum amount of sample material as possible with explanation of calculations based on averages and clear troubleshooting plan
PS: I am short of reputation to add important tags to enhance searching, e.g.: ELISA, validation