A broad interpretation of the question
The question refers to the “folding” of proteins. This term — and the associated biological problem — is often used in the rather specific sense of the attainment of a structure with a particular thermodynamic minimum free energy. I take the intent of the OP (original poster) to be whether, or to what extent, one component of a polyprotein influences the structure adopted by another by physical association. I shall extend the question to that as I think an attempt to answer it will be more interesting and rewarding.
The general and the specific
I do not think one can assume that there will be a single answer to the question that will be true for all polyproteins. Nature is seldom so obliging, and there is no guarantee that what may be true for a polyprotein translated from a viral RNA will also be true for one translated from a cellular RNA. Even within these categories it would be prudent to consider things on a case by case basis.
The answer in summary
Comparisons of the sort suggested have been made in certain cases or are implied by the accumulated work in others. In the polyproteins of both RNA virus and cellular biosynthetic complexes the evidence suggest independent folding in many cases. However in some viral polyproteins, particular pairs of protein domains may be have as a single folded protein in precursors to proteolytically cleaved subsequent forms.
Viral polyproteins
Comparisons of the sort suggested by the OP have, in fact, been performed, although not on complete viral polyproteins — rather restricted to examining possible interactions of particular subsections of the latter. I can see two reasons for this:
Practical Difficulty. I am not aware of any viral polyprotein that has had its total structure determined. This may be because large protein assemblies are generally difficult to crystalize, or because the flexibility of the linkers may mean most do not adopt a single structure, or because even if they do they may not maintain it for any length of time before proteolysis occurs.
Insufficient interest in the question. I suggest that most virologists would assume that the sole or primary reason for synthesis of RNA virus proteins as a polyprotein is that the mechanism of eukaryotic translational initiation precludes polycistronic mRNAs (in all but a few cases not relevant here), and the way the linkers space the protein domains suggests that they fold independently of one another. Furthermore, the unrelated nature of many adjacent protein domains (polymerases, proteases, structural proteins) — and the non-conserved organization of neighbours — gives no reason to suppose that they interact in any way. There is a long history of the crystallization of individual RNA viral proteins expressed from artificial constructs including only the open reading frame of the viral RNA, as exemplified most recently by the expression of Covid proteins for research and vaccine development. This also suggests that individual proteins fold independently.
Comparison studies on partial viral polyproteins
Nevertheless, it has been recognized that — and there has been considerable interest in the fact — the viral polyprotein system has been refined and adapted to the infection. In some cases, for example, there is a process of protein maturation by successive proteolysis, with intermediates having functionally different properties to the mature end-product. For this and other reasons structural analysis and comparisons of parts of the polyprotein have been performed with different viruses and proteins, some of which were reviewed by [Yost and Marcotrigiano in 2013].(https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3660988/)
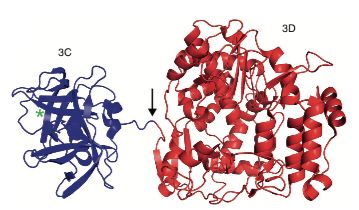
One example (from this review) is the 3C/3D precursor of poliovirus, in which the two components are well separated (above) and, to quote, “Overall, the structures of 3C and 3D alone are very similar to their counterparts in the 3CD precursor.“
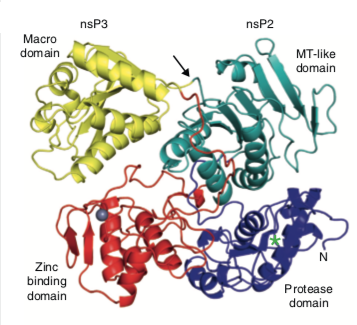
The preceding result may be the one anticipated, but the nsP2/nsP3 precursor of Sindbis virus illustrates a quite different situation. Here the two proteins components interact strongly in the precursor (below), suggesting that after proteolysis a rearrangement of the structures or interaction with some other protein might occur. Suffice it to say that the situation is complex and remains to be resolved. However a comparison of with the structures predicted by alphaFold might be informative.
Cellular Polyproteins
The situation with cellular polyproteins is rather different. These relate synthetic pathways in which the enzymes catalysing successive steps are organized into a pipeline. In the (relatively few) examples the eukaryotic version is a polyprotein, whereas the prokaryotic equivalent is a multi-enzyme complex of similar, but distinct, components. In contrast to the viral polyproteins, there seems no a priori reason for a polyprotein in this case, and, indeed, this is not subject to (natural) proteolytic cleavage.
Polyproteins of this type have been crystallized only relatively recently, although there are several structures of individual components and prokaryotic equivalents. I shall consider two examples.
The first is enzymes of the shikimate pathway, responsible for the synthesis of aromatic amino acids. In bacteria, such as Escherichia coli, this comprises seven individual enzymes, but in lower eukaryotes such as Neurospora crassa a polyprotein of five of these, the arom complex, is employed. (Higher animals lack the ability to make aromatic amino acids, and plants use separate enzymes encoded by the chloroplast genome.) The structure of the arom complex polyprotein was determined for the thermophilic fungus, Chaetomium thermophilum, by Verasztó et al. A feature of this polyprotein is that the individual enzyme domains move and function independently of one another with little effect from their linkages. Although many structures have been reported for the individual enzymes of the complex, these differ between species, and I can find none for C. thermophilum in the protein data bank. Hence the exact test proposed by the OP has not been carried out, but the indirect evidence suggests independent folding. A structure, from Candida albicans, has subsequently been reported.
The second example of a cellular polyprotein is the enzymes of the fatty acid synthase pathway in mammals, and the homologous polyketide synthases in bacteria (only few of which synthesize fatty acids). The first such structure was reported by Maier et al. in 2008. The porcine complex is shown below, and has a more open and flexible design than the arom complex, that seems to have allowed insertion and deletion of different catalytic domains during evolution. Again, this argues in favour of independent folding of individual components, although I am not aware of direct comparison with individually expressed fragments. I suspect that there might be structures available to do this, although I haven’t had the time to examine this further myself.
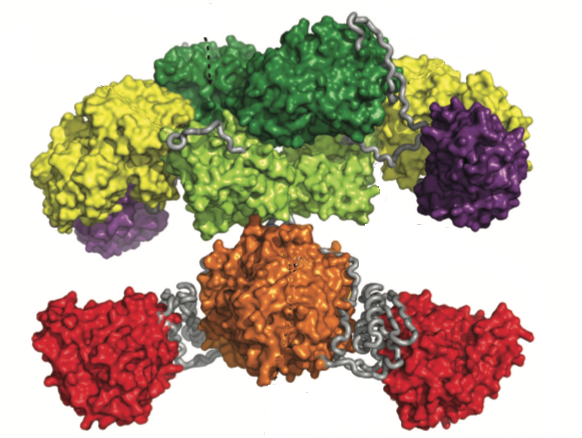
I would conclude by noting that although the individual components of these cellular polyproteins interact with one another to cooperate in the biosynthesis, the flexibility they exhibit allows transfer of the growing substrate from one component to another.
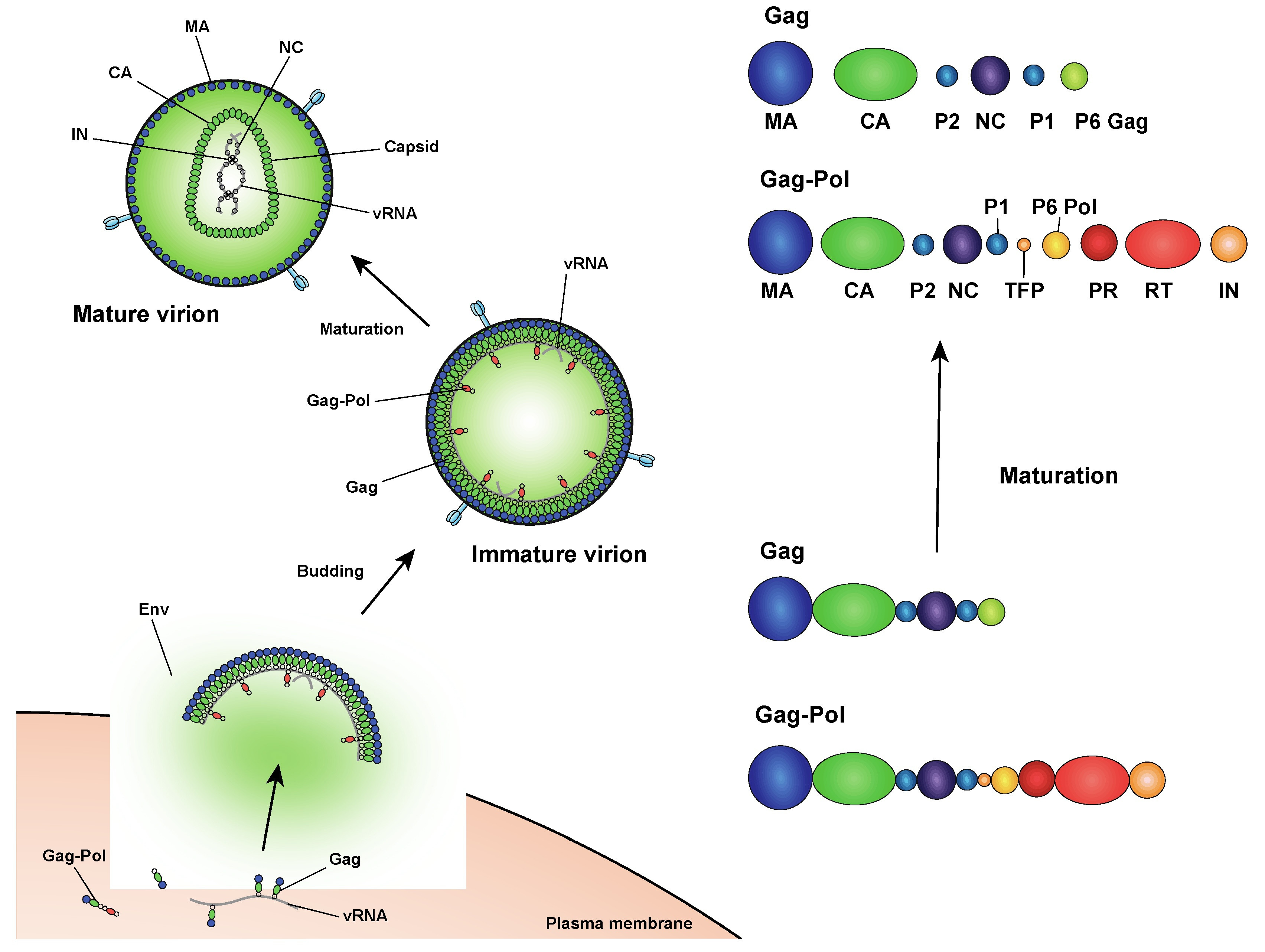 This question is not a mere curiosity. Many proteins have multiple globular domains which makes structure determination difficult because these domains are very loosely joined together. It would be much easier if we can solve the globular domains individually instead of solving the whole protein at once.
This question is not a mere curiosity. Many proteins have multiple globular domains which makes structure determination difficult because these domains are very loosely joined together. It would be much easier if we can solve the globular domains individually instead of solving the whole protein at once.