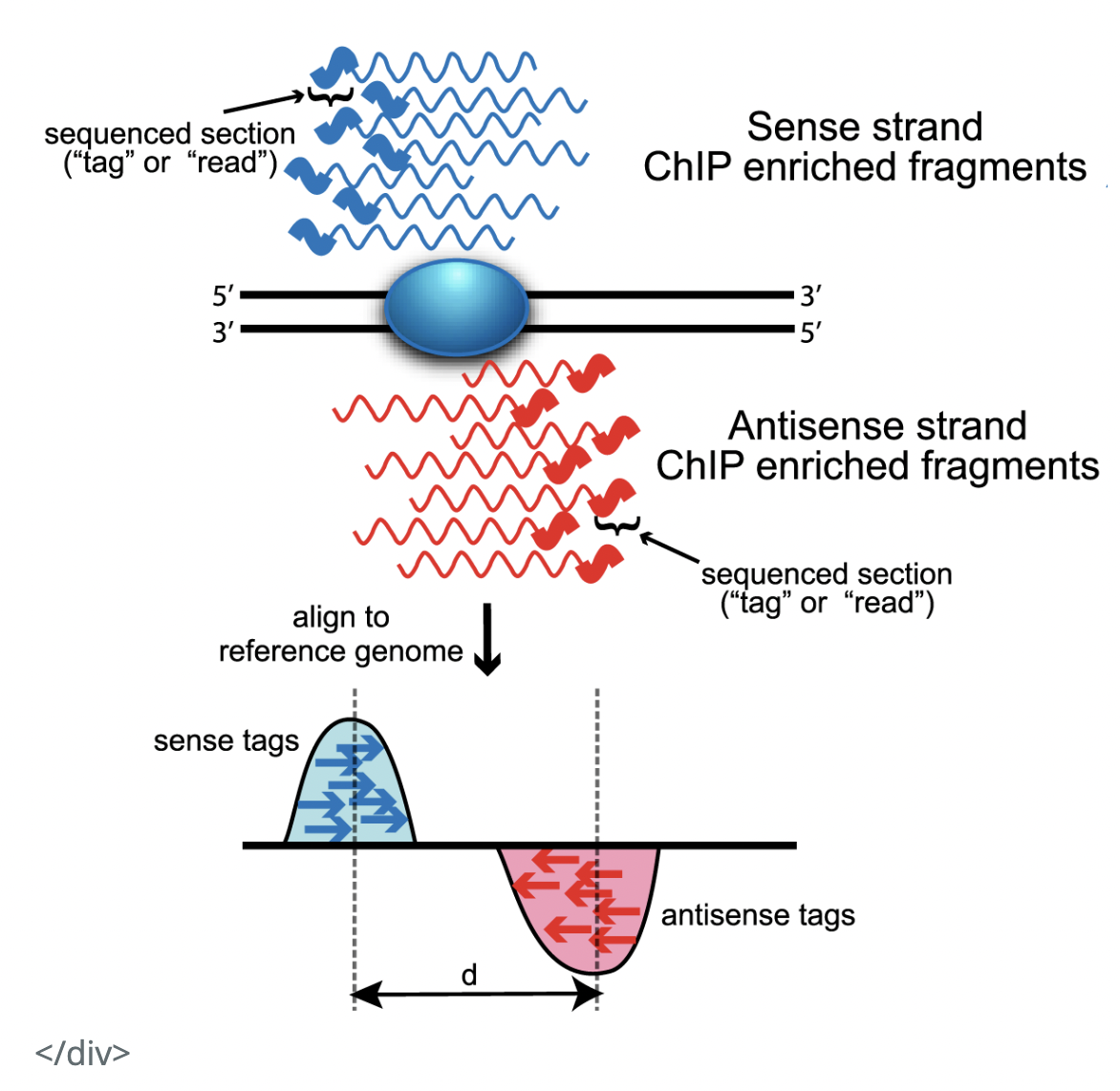
I had a few questions raised by the below diagram in relation to how ChIP-seq reads look like (source). I would be very grateful for your insight on them!
From the below diagram, it seems that, in a ChIP-seq experiment, we get information about the binding site in relation to both the Watson and Crick strands. That is, from our experiment, we get reads complimentary to both strands.
(Q1) I suppose this is because proteins bind to both strands of DNA, so that in our ChIP-seq experiment, we ultimately sequence reads from both the Watson and the Crick strands? Is that true?
Also, my impression from the MACS paper was that "ChIP-Seq tags represent only the ends of the ChIP fragments, instead of precise protein-DNA binding site" (Background).
(Q2) Therefore, shouldn't it be that we get reads mapping to either side of the binding site on BOTH strands? That is, we get reads more towards the 3' end of the Watson strand, and reads more towards the 5' end of the Watson strand, and similarly for the Crick strand.
(Q3) The diagram below shows we only get reads more towards the 5' end of the strands. Why is that?
(Q4) Finally, I know that MACs shifts reads by some bases towards the 3' end for each strand, in order to interpret the precise binding site location. It seems to assume what I am asking about in (Q3). However, if the answer to my (Q2) is "yes, we do get 4 such clusters of reads", then why doesn't MACs account for that?