TLDR; Answer: You could consider this particular residue to belong to both structural elements, but it's a tricky call and depends on the method of secondary structure assignment.
Ambiguous secondary structure allocation comes up fairly often. Whilst obviously, not many people will be able to use this protein specifically, the below approach could be useful for other proteins.
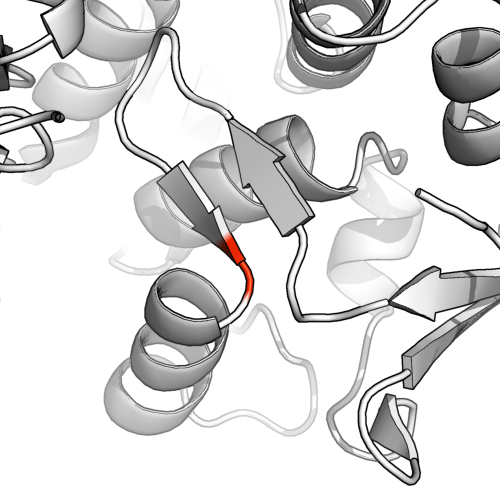
Default pymol assignment
In PyMol I used fetch 1ae9 and highlighted M at position 255 on the A chain in red. I can see why this isn't a satisfying representation: the beta sheet allocation is very short, and the residue in question is clearly the beginning of a helix.
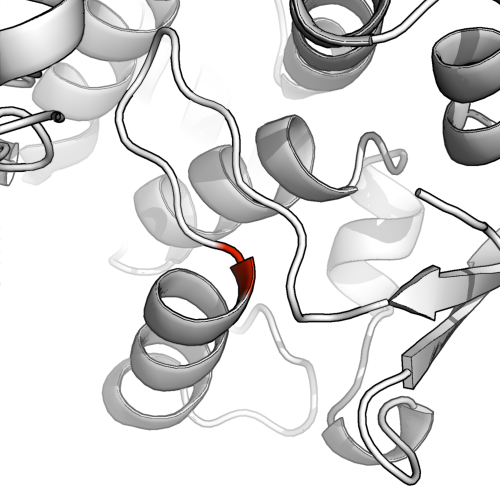
DSS is more conservative
To run a more conservative secondary structure allocation run dss in pymol (watered down dssp). This reveals that the beta sheets were actually quite putative.
We can see why when we look at the stick model (below). There are only a few (2-4) residue pairs in proximity that would be available for hydrogen bonding, even assuming all 4 form H-bonds, calling this a bona fide beta sheet could be seen as a bit generous.
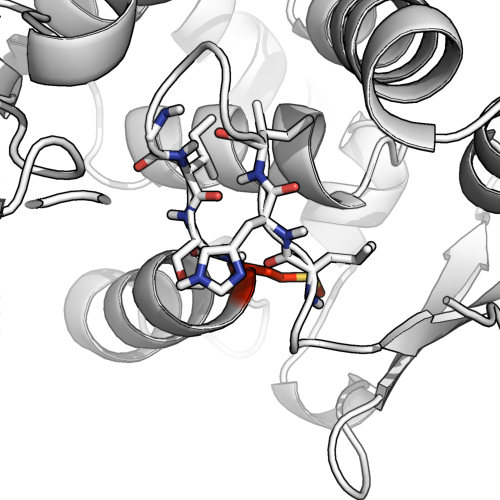
Dual assignment can be okay.
Looking at this example (1ae9), we see the potential for H-bonding with another putative beta sheet, as well as a backbone angle that starts to form a helix. It's a classic case of ambiguous dual assignment. @AlexanderDScouras draws a reasonable conclusion that both is okay and to directly answer the question: dual assignment is possible, and allowed. I would prefer to rule out beta sheets in this particular case, but on the condition that one highlights that there is plenty of hydrogen bonding in the hairpin loop.
So long as it's reasonable.
So long as you can reasonably justify the assignment it's probably okay. You can manually assign any residue and secondary structure.
# set residue 155 to be alpha-helical
alter 155/, ss='H'
# update the scene in PyMOL to reflect the changes.
rebuild
Evidence is always a good idea to support manual assignments. A quick and thorough way would be to run the sequence through a few secondary structure predictors, or use the genesilico metaserver to save yourself some hassle (you'll need an account) (this does feel like going back a step since you now have a structure, so exercise caution - if the sequence prediction doesn't look correct according to the structure, it's probably not). Another method as suggested by shigeta is running this through a Ramachandran plot (RAMPAGE is a favourite).
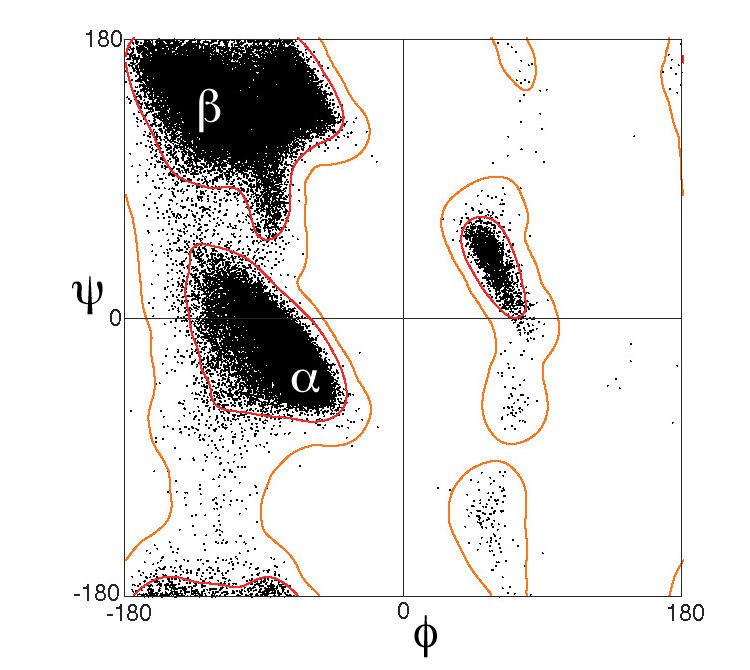
If it's very very unclear then discuss it thoroughly and clearly whenever this part of the protein is relevant. These secondary structure assignments have somewhat arbitrary cut-offs after-all, and when things get near the thresholds, it's important to approach the situation with clarity and specificity.
Ramachandran plot image source: By Dcrjsr - Own work, CC BY 3.0, https://commons.wikimedia.org/w/index.php?curid=9105708